
Hemophagocytic lymphohistiocytosis (HLH)
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening disorder characterized by pathologic immune activation and systemic hyperinflammation.
The inherited form of HLH is known as primary hemophagocytic lymphohistiocytosis (pHLH). pHLH is a rare disorder associated with impaired cytotoxic function of NK cells and CD8+ T cells. It typically manifests during infancy and is invariably fatal if untreated.
Other forms of HLH are known as secondary HLH, including a subset referred to as macrophage activation syndrome (MAS). HLH/MAS is a rare, potentially fatal complication of rheumatological conditions, especially systemic juvenile idiopathic arthritis (sJIA) and adult-onset Still's disease (AOSD), that can result in significant morbidity to both pediatric and adult patients.

HLH publications
The publications on this page are sponsored by Sobi and pertain to select Sobi products in this disease area. This page is not intended to serve as a comprehensive overview of all publications in this disease area. Publications may include information for which the product is not approved and/or that is inconsistent with the product uses described in the Prescribing Information. Sobi does not suggest or recommend the use of Sobi products in any manner other than as described in the Prescribing Information.
HLH resources
Review the recommended resources for additional clinical insights.
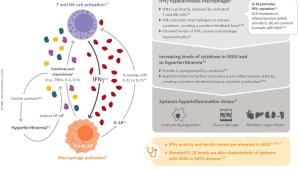
View the infographic to learn more about the role of IFNg in MAS pathophysiology.
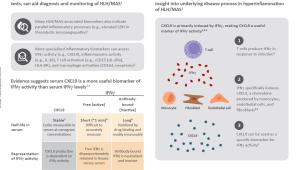
View the infographic to learn more about the latest biomarkers for MAS management.

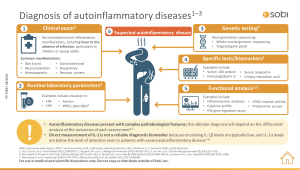
View the infographic to learn more about the diagnosis of MAS.

View the infographic to learn more about treatment aims and considerations in MAS management.
Neonatal-onset multisystem inflammatory disease (NOMID)
NOMID is a rare, predominantly pediatric autoinflammatory disease that can be challenging to diagnose and can cause long-term disability without treatment. Globally, only 100 people have been reported to have NOMID.

NOMID publications
The publications on this page are sponsored by Sobi and pertain to select Sobi products in this disease area. This page is not intended to serve as a comprehensive overview of all publications in this disease area. Publications may include information for which the product is not approved and/or that is inconsistent with the product uses described in the Prescribing Information. Sobi does not suggest or recommend the use of Sobi products in any manner other than as described in the Prescribing Information.
NOMID resources
Review the recommended resources for additional clinical insights.


Deficiency of the interleukin-1 receptor antagonist (DIRA)
DIRA is an ultra-rare, autosomal recessive disorder caused by mutations in the IL1RN gene, leading to a loss of production and function of IL-1Ra. DIRA generally presents at or near birth and can escalate to life-threatening inflammation. To date, DIRA has been identified in fewer than 50 people worldwide.

DIRA publications and case reports
The publications on this page are sponsored by Sobi and pertain to select Sobi products in this disease area. This page is not intended to serve as a comprehensive overview of all publications in this disease area. Publications may include information for which the product is not approved and/or that is inconsistent with the product uses described in the Prescribing Information. Sobi does not suggest or recommend the use of Sobi products in any manner other than as described in the Prescribing Information.
DIRA resources
Review the recommended resources for additional clinical insights.
Hereditary tyrosinemia type 1 (HT-1)
HT-1 is a rare, autosomal recessive genetic metabolic disorder with a global birth incidence of 1/100,000 in most areas. HT-1 is caused by a deficiency in the enzyme fumarylacetoacetate hydrolase (FAH), which is essential for the final breakdown of the amino acid tyrosine. Without proper breakdown, tyrosine and its metabolites build up in the liver, potentially resulting in severe liver disease. Tyrosine may also accumulate in the kidneys and central nervous system.

We don’t have any documents related to this therapeutic area at this time.
Connect with an MSL
Sobi Medical Science Liaisons are highly trained, scientific and clinical subject matter experts who can provide in-depth answers to your disease and treatment questions.